Thorough Testing of Internal Reference Scaling (IRS)¶
Phillip Wilmarth, OHSU¶
January 5, 2019¶
What and Why¶
This notebook will:¶
- give a brief overview of Internal Reference Scaling
- why we need IRS and what IRS corrects
- IRS algorithm specifics
- IRS experimental design using reference channels
- show that 4 commonly used normalization methods do not work
You will learn:¶
- normalization between TMT plexes is fundamentally different
- MS2 sampling effect has to be removed
- working with data in natural scales has major benefits
What is IRS?¶
The problem¶
If we label the same sample in more than one channel in an isobaric labeling experiment (one labeling kit of samples will be called a "plex" to avoid confusion with "run" or "experiment") and perform some very basic data normalizations, we will see that the measurement values for those channels at any level (PSM, peptide, or protein) are very nearly identical. We can aggregate (sum) the data (intensities or peak heights) from the individual PSMs into peptide totals or into protein totals (based on protein inference rules). It does not matter if the single plex was analyzed using a single LC run or multiple LC runs (fractions). Data quality improves as a function of aggregation with protein level data having lower variation than peptide or PSM data (see this notebook). Different channels of the same sample (technical replicates) can be used to verify that data aggregation steps do not distort the inherent realtive precision of isobaric measurements between channels in a single plex.
What happens when we have the same samples in different plexes? Even with 11 channels currently avaialble, more channels may be needed to accommodate all of the biological samples and more than one isobaric plex will need to be used. If we do basic single factor normalizations (matching median intensities or total intensities), we will find that we do not get similar intensity measurements of the same thing between plexes. The overall intensity of reporter ions depends not only on the abundance of the peptide, but on when the peptide was "sampled". The fragmentation scans that provide the peptide sequence and the reporter ion signals (this can be a single scan or a pair of linked scans) are a few 10s to 100s of milliseconds in duration. The extracted ion chromatograms (XIC) of peptides are most often 10s of seconds (30 seconds to 1 minute are typical). If the fragmentation scan occurs at the apex of the XIC, the report ions will be more intense that from fragmentation scans taken at other points along the XIC (such as near the baseline).
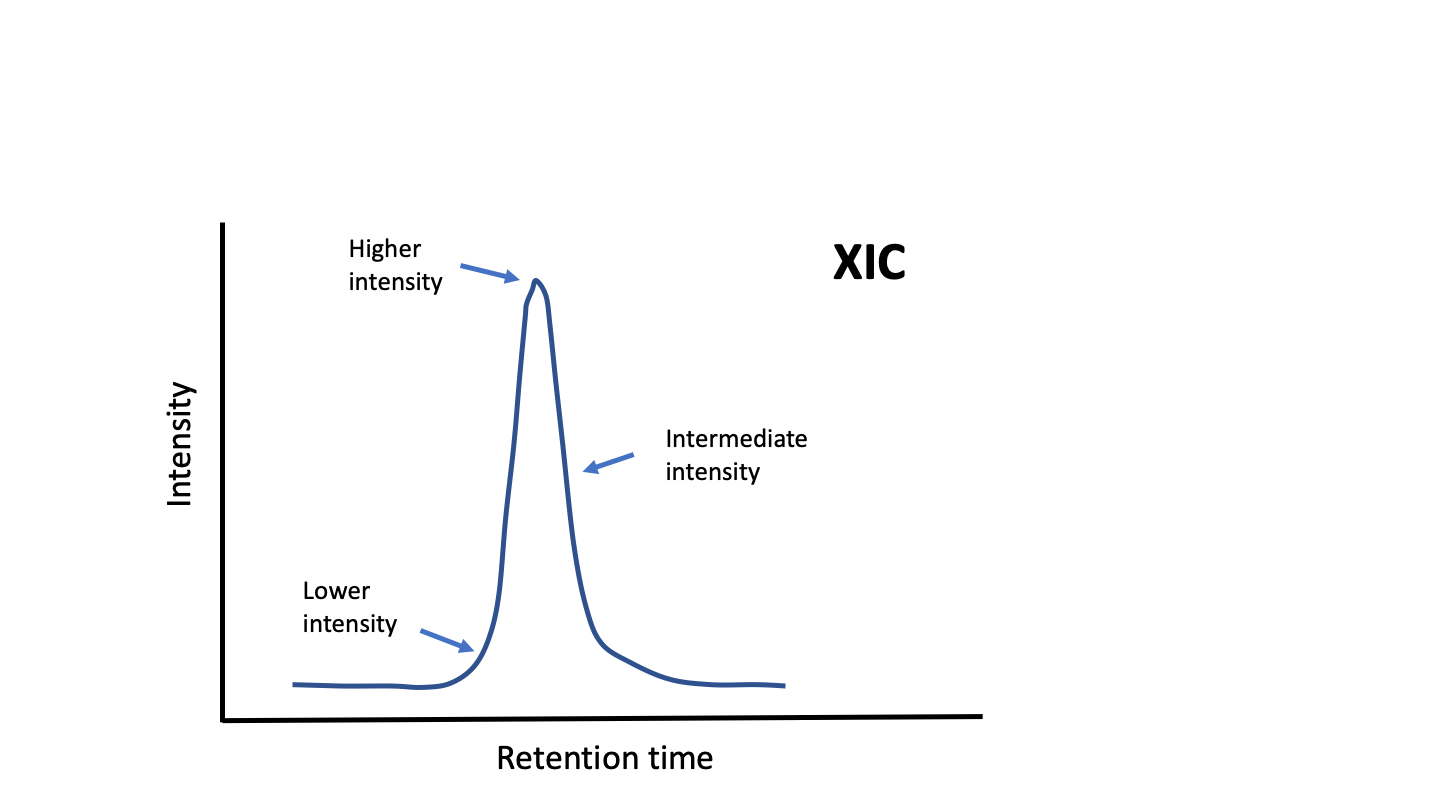
Isobaric labeling¶
Isobaric labeling (Ref-1, Ref-2) is a precise relative quantification method because reporter ion intensities are simultaneously measured in a single instrument scan. (Isobaric tag and TMT will be used interchangeably.) Shotgun proteomics works by chopping proteins into large numbers of peptides that can be separated by liquid chromatography and sequenced by mass spectrometers. The overwhelmingly large number of measurements from all of the small pieces is far less useful than aggregating the data from the pieces (the peptides) back into the whole (the proteins).
The relative precision of the reporter ions is maintained when the data from the pieces are summed together. The intensities of the pieces will vary depending on many factors such as analyte abundance, and when the analyte is sampled during its elution profile. Isobaric labeling reporter ions come from MS2 "snapshots" of the eluting peptide. MS2 scan selection in shotgun proteomics is kind of a random process (Ref-3) given the sample complexity. MS2 scans are not like integrated MS1 peak heights or areas, which tend to be more stable values. MS2 scans taken from the leading or trailing edge near the baseline will be less intense than MS2 scans taken near the chromatography peak apex. The intensities of ions in MS2 scans (including the reporter ions) are highly variable.
Within a single TMT plex (one set of isobaric labeling tags, a.k.a. one kit), the random nature of MS2 scan selection can be pretty safely ignored. Pretty much any way that data can be analyzed ends up working okay because of the relative precision of the reporter ions within each scan. The situation changes dramatically when data from more than one TMT plex is combined in larger experiments. Now the intensity differences that depend on when the MS2 scans were selected in each TMT experiment will dramatically alter the scales of the intensity values, and the data cannot be combined until these differences are removed.
Need for reference channels¶
Ironically, in much the same way that the similarities between reporter ions within the same scan are retained during data aggregations for a single TMT plex, the differences between reporter ions from different MS2 scans persists after data aggregations and conventional normalization methods. A specific algorithm to determine and correct the MS2 sampling effect has to be used.
Adding a common reference channel to each plex has been done in isobaric labeling for many years. The reference channel has mostly been used as a common denominator in ratios with each biological sample. This has the desired effect of normalizing the ratios to a common scale. The downside is that ratios are not very intuitive, have compressed dynamic range, have half of the values between 0 and 1.0 and the other half between 1.0 and infinity, and are not tolerant of zero values (missing data). Because of that asymmetry, logarithms are often necessary, and those further compress the dynamic range of the measurements are even less intuitive that ratios.
Another drawback with ratios is that they are like the edges in a network. Their number grows much more rapidly that the number of network nodes. In an isobaric labeling study with a single common channel, there is no ambiguity in taking ratios of each channel to the reference. There is only one way to do that. If you have half of the channels as controls and half as treatments, then there is ambiguity about what ratios to form to compare control to treatment. If you have 2 channels (one of each condition), there is one ratio. If you have 2 of each condition, there are 4 different ratios. If you have 3 of each condition, there are 9 ratios. The number of ratios is the product of the number of samples in each condition. 10-plex TMT could accommodate 5 of each condition and result in 25 ways to take ratios. Statistically (mathematically) this is not good. It is an over-determined problem where there are more ratios than degrees of freedom.
IRS method maintains the natural measurement scale¶
Is there another way to make use of reference channels to put reporter ion intensities on a common scale that does not involve ratios? In protein expression studies using TMT, a large number of PSM data points will aggregate into a much more manageable and shorter list of proteins. The reference channels, being the same in each TMT plex are like yardsticks (or meter sticks) that measure an observed value for each protein in each plex. The main assumption of IRS is that those numbers should have been the same since the reference channel was the same. The measured values are not identical in each plex (mostly) because of random MS2 scan selection. We can mathematically make them all the same between the plexes using scaling factors for each protein, as shown in the diagram below.
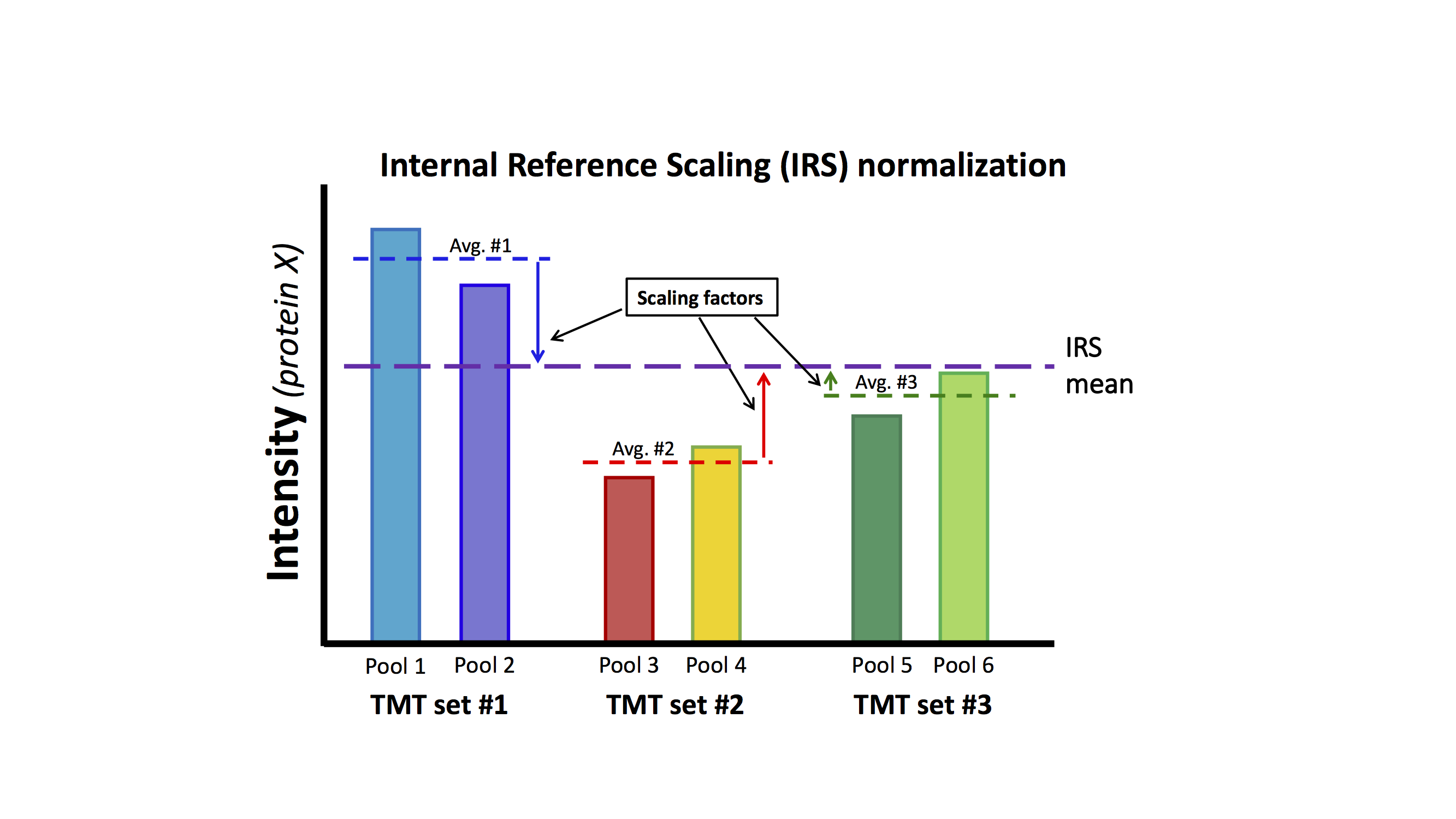
Since the non-reference channel(s) in each single plex have high relative precision that is independent of the magnitude of the intensity values, we can scale all of these other channels by the same scale factors that we are using on the reference channels. This adjusts all channels in each TMT plex on to a common intensity scale. This method is called internal reference scaling (IRS) and was first described in (Ref-4). After IRS, any channel can be directly compared to any other channel (within TMT plex and between TMT plexes become equivalent). Importantly, the data used for the scaling factors are independent of the data used in statistical testing.
It is time for a conceptual description of IRS using jazz hands. Think of each plex's channels as the fingers on a hand. If we have an experiment with two plexes, that is like our two hands. Because of pseudo random MS2 sampling we have different overall intensity scales. This is like our two hands being at different heights. The IRS method measures the heights of the hands and determines how much to move one hand down and the other hand up to bring them into alignment. The fingers (hopefully) all move with the hands, so when the hands are in alignment, then all of the fingers are also in alignment. That is all that there is to IRS. Really.
The IRS experimental design outlined in (Ref-4) was to use duplicate channels of the same pooled internal standard in each TMT plex. With only 10 or 11 channels per plex available, this might seem excessive. However, there are several quality control and accuracy arguments to support two channels instead of just one. The average of two measurements will be much better than just one, and everything hangs on the accuracy of the scaling factors. It is possible (but hopefully rare) to accidently mis-label samples and it may not be clear which channels where the standards. The strong similarity of the two duplicate standard channels can be used to find them in the presence of the biological samples, which are not usually as similar to each other (see this notebook).
Testing and validating IRS¶
Validation of IRS was done in the Supplemental materials of Ref-4 by using half of the pooled standards for IRS and using the other half as a validation set. A recently completed experiment used seven 11-plex TMT labelings to study exosomes from 3 groups with 20 biological samples each. Each 11-plex TMT experiment used duplicated pooled standard channels. That allowed 9 channels for biological samples per plex. Seven plexes would have 63 available channels. After the 60 biological samples, that left 3 additional channels. Extra pooled standard samples were labeled and analyzed in the three extra channels. Those 3 extra channels were distributed in three of the 7 TMT experiments.
This allows a rigorous independent validation of the IRS method. There are three TMT experiments with triplicate pooled standards run in the same TMT experiment. We also have the three extra pooled standards in different TMT plexes that can be compared before and after a proper IRS normalization based on pairs of pooled standards. Remember that the master pooled standard protein mixture is created from equal amounts of all 60 samples and each pooled standard channel is an independent digestion and labeling of an aliquot of the master pooled standard protein mixture.
Data description¶
The data are from human urine exosomes from the Christina Binder lab at OHSU. Exosome isolation was performed at Ymir Genomics (Boston, MA) by Shannon Pendergrast. eFASP digestion (Ref-5), 11-plex TMT labeling (Thermo Fisher Scientific), liquid chromatography, and mass spectrometry analysis was performed at OHSU by Ashok Reddy. The IRS experimental study design was used to accommodate 20 samples per condition in 7 TMT plexes. 30-fraction online high pH RP/low pH RP LC separations were done. The SPS MS3 method (Ref-6) was used on a Thermo Fusion with the manufacturer's default method.
Data analysis was performed by Phil Wilmarth, OHSU. RAW files were converted to text files using MSConvert (Ref-7). Python scripts created MS2 spectra for database searching and tables of the reporter ion intensities (peak heights) for each MS3 scan. Comet database searches (Ref-8) were performed using a canonical protein database (downloaded with software available here), tryptic cleavage, a wider parent ion mass tolerance search, static TMT label modifications, and variable oxidation of methionine. An extended version of the PAW pipeline (Ref-9) was used for filtering of PSM identifications by accurate masses and by the target/decoy strategy (Ref-10) to obtain 1% PSM FDR. Parsimony (Ref-11) and extended parsimony analyses were used to produce a final list of identified proteins and to determine which peptides were unique to the final protein groups. Total protein intensities were computed as the sums of all unique peptide reporter ion signals (Ref-12). IRS normalization of the TMT data across the 7 TMT experiments was done using Python scripts. The analysis presented here was performed with a Jupyter notebook and an R kernel.
References¶
Ref-1. Thompson, A., Schäfer, J., Kuhn, K., Kienle, S., Schwarz, J., Schmidt, G., Neumann, T. and Hamon, C., 2003. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Analytical chemistry, 75(8), pp.1895-1904.
Ref-2. Ross, P.L., Huang, Y.N., Marchese, J.N., Williamson, B., Parker, K., Hattan, S., Khainovski, N., Pillai, S., Dey, S., Daniels, S. and Purkayastha, S., 2004. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Molecular & cellular proteomics, 3(12), pp.1154-1169.
Ref-3. Liu, H., Sadygov, R.G. and Yates, J.R., 2004. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Analytical chemistry, 76(14), pp.4193-4201.
Ref-4 Plubell, D.L., Wilmarth, P.A., Zhao, Y., Fenton, A.M., Minnier, J., Reddy, A.P., Klimek, J., Yang, X., David, L.L. and Pamir, N., 2017. Extended multiplexing of TMT labeling reveals age and high fat diet specific proteome changes in mouse epididymal adipose tissue. Molecular & Cellular Proteomics, 16(5), pp.873-890.
Ref-5 Erde, J., Loo, R.R.O. and Loo, J.A., 2014. Enhanced FASP (eFASP) to increase proteome coverage and sample recovery for quantitative proteomic experiments. Journal of proteome research, 13(4), pp.1885-1895.
Ref-6 McAlister, G.C., Nusinow, D.P., Jedrychowski, M.P., Wühr, M., Huttlin, E.L., Erickson, B.K., Rad, R., Haas, W. and Gygi, S.P., 2014. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Analytical chemistry, 86(14), pp.7150-7158.
Ref-7 Kessner, D., Chambers, M., Burke, R., Agus, D. and Mallick, P., 2008. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics, 24(21), pp.2534-2536.
Ref-8 Eng, J.K., Jahan, T.A. and Hoopmann, M.R., 2013. Comet: an open‐source MS/MS sequence database search tool. Proteomics, 13(1), pp.22-24.
Ref-9 Wilmarth, P.A., Riviere, M.A. and David, L.L., 2009. Techniques for accurate protein identification in shotgun proteomic studies of human, mouse, bovine, and chicken lenses. Journal of ocular biology, diseases, and informatics, 2(4), pp.223-234.
Ref-10 Elias, J.E. and Gygi, S.P., 2007. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature methods, 4(3), p.207.
Ref-11 Nesvizhskii, A.I. and Aebersold, R., 2005. Interpretation of shotgun proteomic data the protein inference problem. Molecular & cellular proteomics, 4(10), pp.1419-1440.
Ref-12 Wenger, C.D., Phanstiel, D.H., Lee, M.V., Bailey, D.J. and Coon, J.J., 2011. COMPASS: A suite of pre‐and post‐search proteomics software tools for OMSSA. Proteomics, 11(6), pp.1064-1074.



































